Introduction
SVOC takes an unannotated VCF file as input and automatically call ANNOVAR to generate necessary annotations, including refGene, dbnsfp47a, and dbscsnv11. Concurrently, SVOC automatically employs TransVar to derive annotations such as transcript, genomic coordinates, transcript-dependent cDNA, protein coordinates, and variant consequence. It utilizes MANE v1.4 (Matched Annotation from NCBI and EMBL-EBI) to annotate canonical transcripts, specifically the MANE Select transcript. However, for certain genes, the MANE Select transcript alone may not be sufficient to report all known variants. This is particularly relevant in genes with mutually exclusive exons, where both exons harbor clinically significant variants. To ensure comprehensive representation, a MANE Plus Clinical transcript is added alongside the MANE Select transcript.
To address the dynamic nature of genomic evidence, SVOC will periodically incorporate updates from these databases and integrate them with the following criteria to determine the final classification.
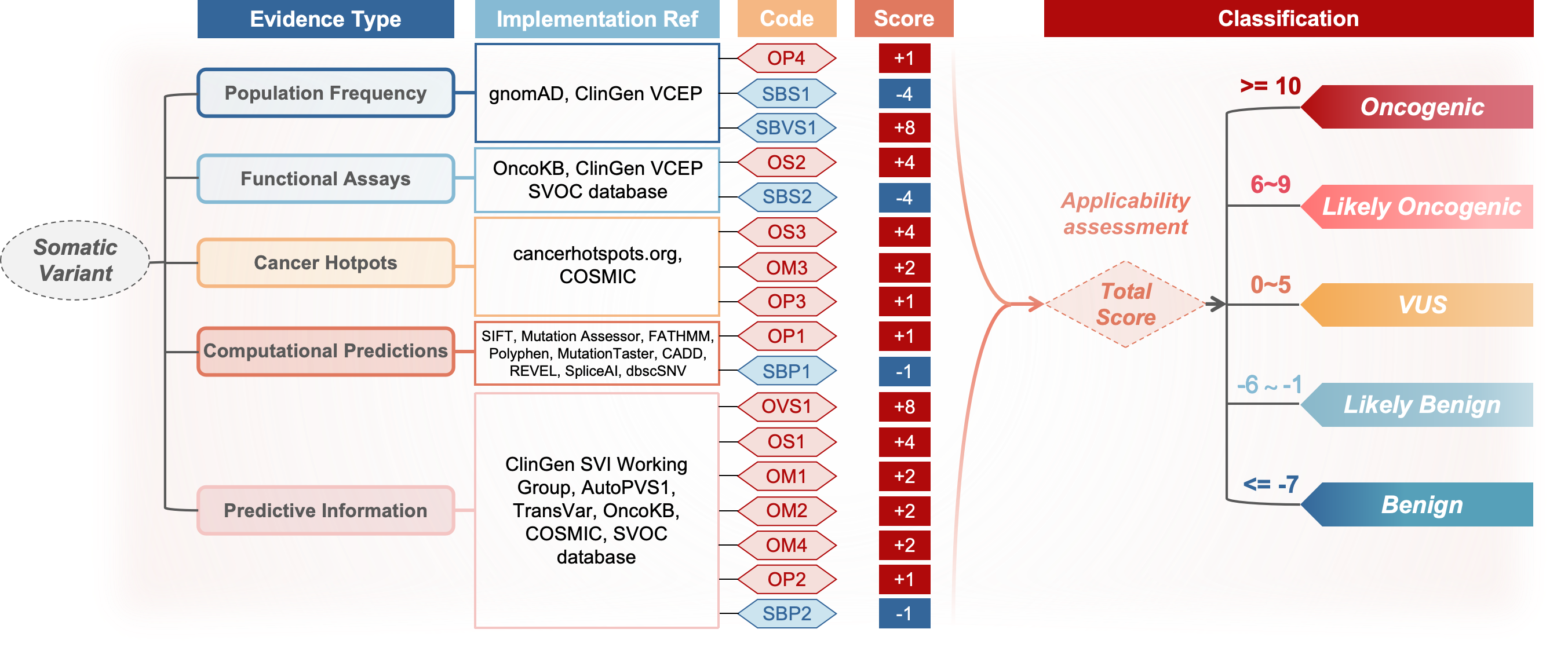
According to the 2022 ClinGen-CGC-VICC SOP, there are five types of evidence to classify the oncogenicity of somatic variants, including population frequency, cancer hotspots, functional assays, predictive information and computational predictions.
Based on these evidence, variants are classified into five categories: Oncogenic, Likely Oncogenic, Variants of Uncertain Significance (VUS), Likely Benign and Benign. There are a total of 17 criteria:
- Population frequency: Somatic Benign Very Strong 1 (SBVS1), Somatic Benign Strong 1 (SBS1) and Oncogenic Supporting 4 (OP4).
- Functional assays: Oncogenic Strong 2 (OS2) and Somatic Benign Strong 2 (SBS2).
- Predictive information: Oncogenic Very Strong 1 (OVS1), Oncogenic Strong 1 (OS1), Oncogenic Moderate 1 (OM1), Oncogenic Moderate 2 (OM2), Oncogenic Moderate 4 (OM4), Oncogenic Supporting 2 (OP2), Somatic Benign Supporting 2 (SBP2).
- Cancer hotspots: Oncogenic Strong 3 (OS3), Oncogenic Moderate 3 (OM3) and Oncogenic Supporting 3 (OP3).
- Computational predictions: Oncogenic Supporting 1 (OP1) and Somatic Benign Supporting 1 (SBP1).
Each criterion has a predefined score from -8 to 8, where higher scores denote greater oncogenicity. Score mappings are: OVS (8), OS (4), OM (2), OP (1), SBVS (-8), SBS (-4), SBP (-1). SVOC automatically scores all 17 criteria and calculates the total score of the variant based on the applicable rules defined in the guidelines (e.g., OM1 cannot be used if OS1 or OS3 is applicable), and assigns a category: O (≥10), LO (6–9), VUS (0–5), LB (-1–-6), B (≤-7).
FAQ
Currently, Single Nucleotide Variants (SNVs) and Insertion/Deletion variants (Indels).
We acknowledge that inversions, gene fusions, and gene expression alterations may play important roles in cancer development and progression. Therefore, when specific guidelines for these types of mutations become available, we will promptly incorporate the corresponding functionalities.
Classification Summary page displays the classification results for user-queried variants.
The Statistics module at the top provides an overview: Donut Plot visually represents the proportion of variants across different oncogenicity classifications; Bar Plot details the variant count per gene and their oncogenicity distribution.
The Table below lists all variants with their location and classification information. The STATUS indicates whether the classification is 'Automated' (system-generated) or 'Adjusted' (manually modified by the user). Use the search box in the table's top-left corner to filter table content globally. To review or adjust how a specific variant meets the 17 classification criteria, click its corresponding Details & Adjust link. This will navigate you to the Classification Details page.
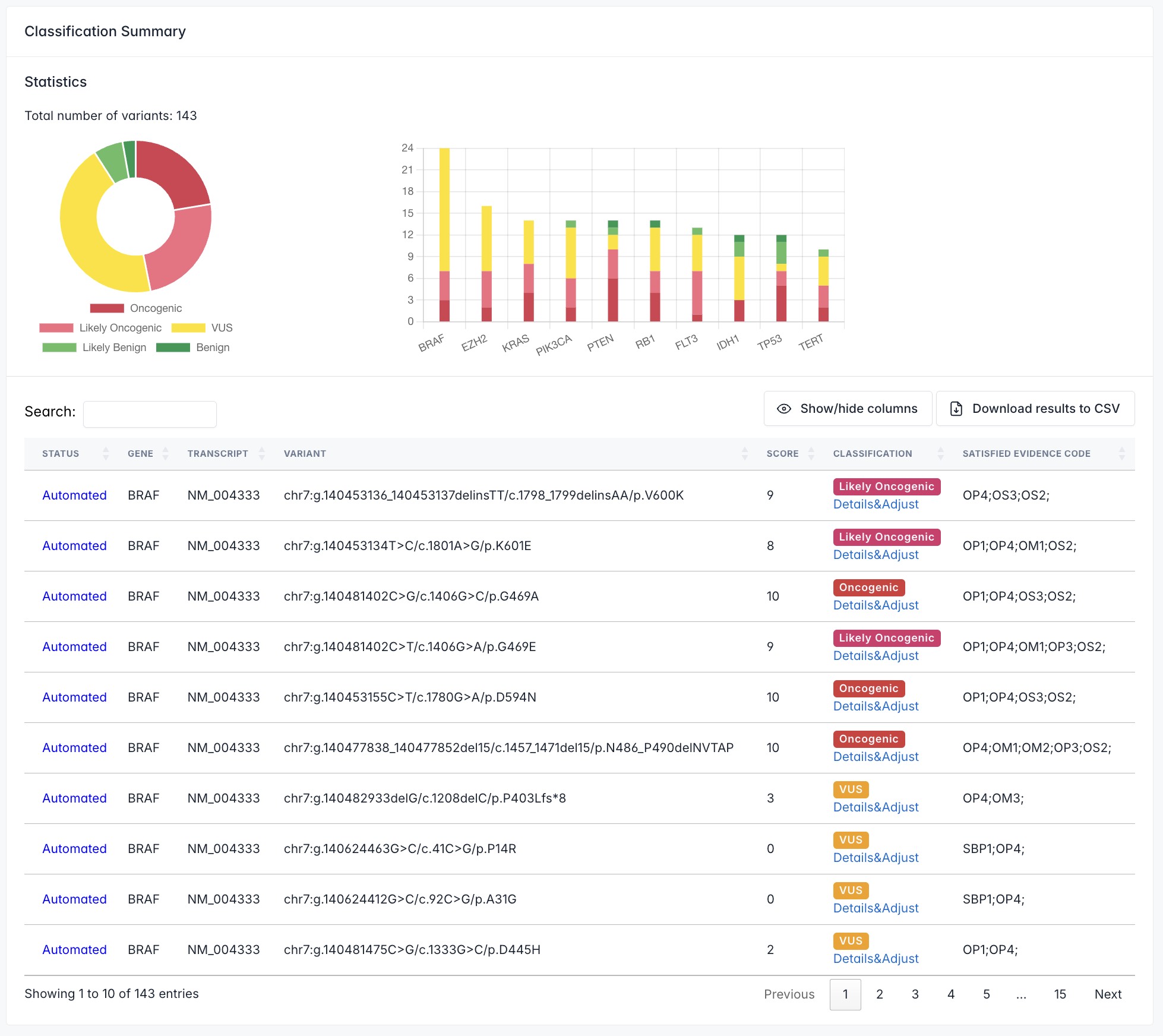
For a detailed description of the evidence codes, please refer to the publication of ClinGen/CGC/VICC guidelines.
SVOC utilizes the canonical transcript from MANE v1.4 (Matched Annotation from NCBI and EMBL-EBI), specifically the MANE Select transcript. However, for certain genes, the MANE Select transcript alone may not be sufficient to report all known variants. This is particularly relevant in genes with mutually exclusive exons, where both exons harbor clinically significant variants. To ensure comprehensive representation, a MANE Plus Clinical transcript is added alongside the MANE Select transcript. For further details, refer to the following resources:
MANE v1.4 with MANE Select for Non-Coding GenesMANE progress update
Classification Details page displays the fulfillment status of the 17 criteria for a specific variant, along with reviewable text evidence for each met criterion. It supports users in interactively refining results using their own internal databases.
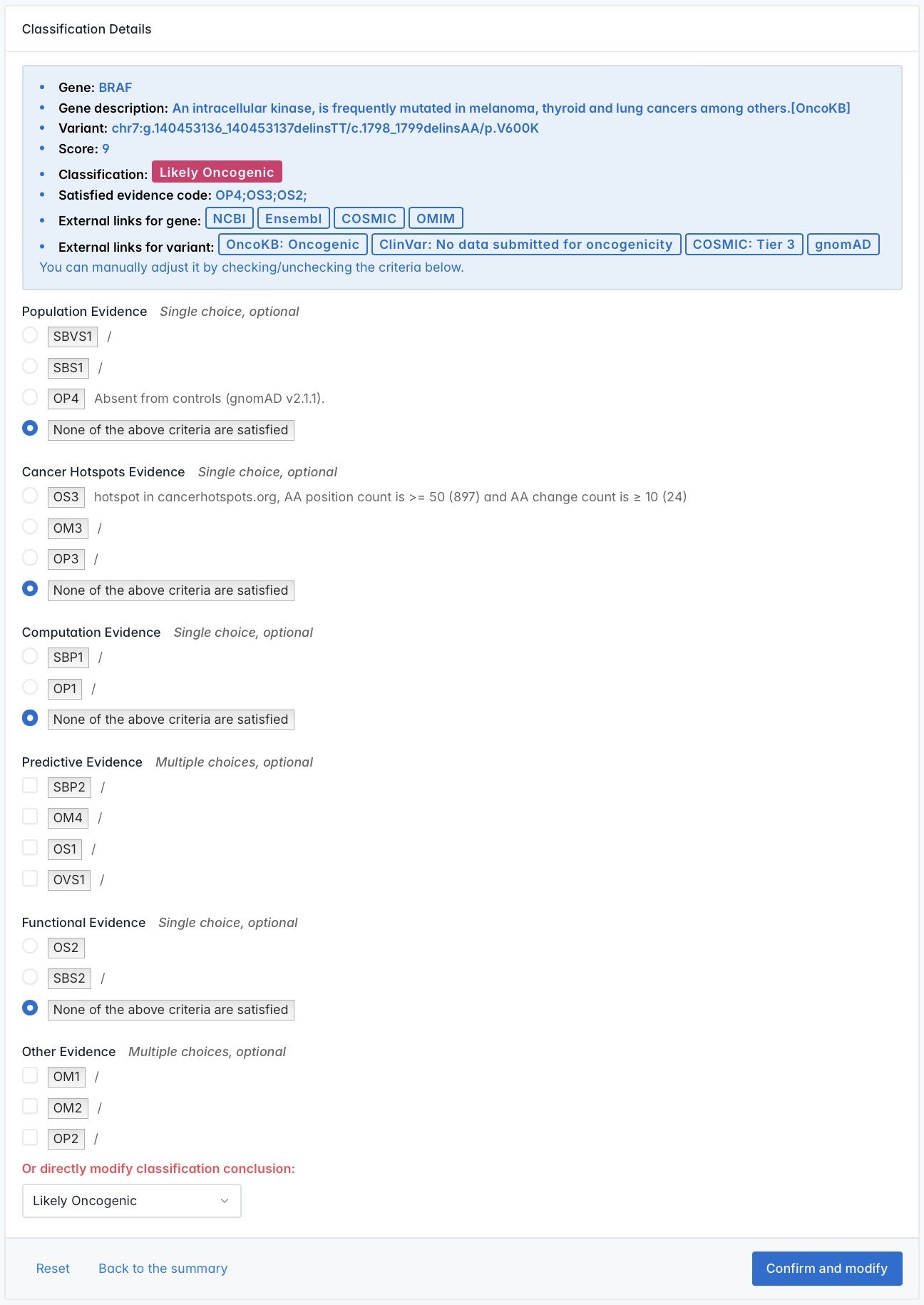
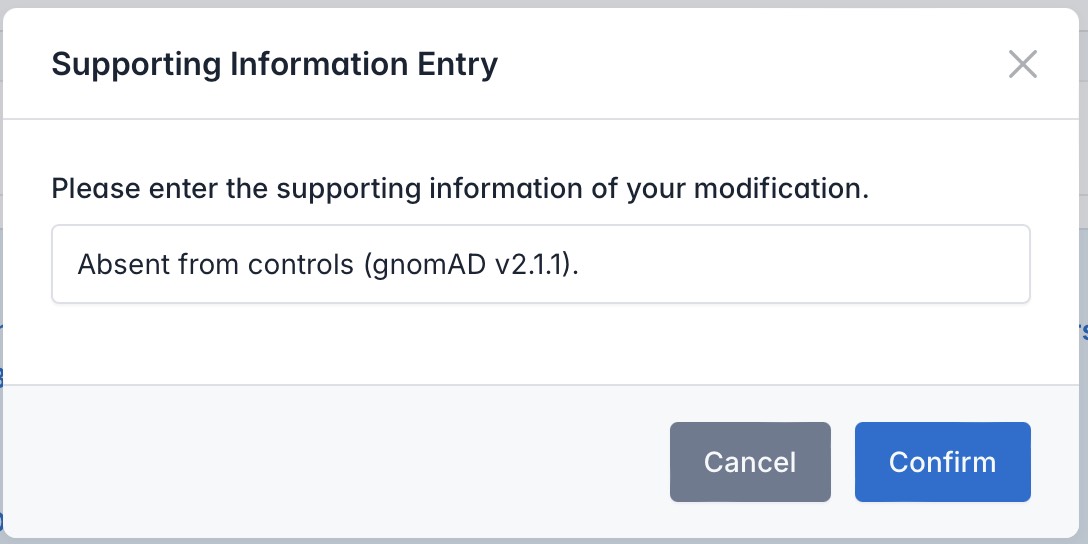
Users can adjust which criteria a variant meets by selecting or deselecting the radio buttons or checkboxes next to each standard. When changes are made, the system will prompt for supporting information. After the user provides and submits this information, the classification result will update automatically.
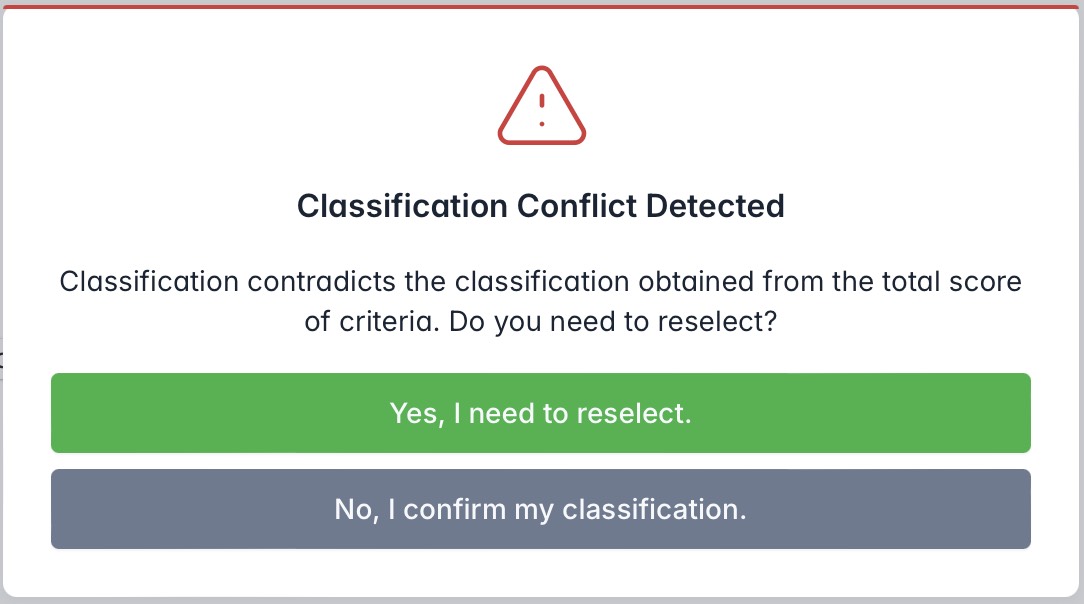
Or, users can directly modify the final classification conclusion using the dropdown menu at the bottom of the page. If the selected classification contradicts the variant's classification based on scoring criteria, the system will prompt the user to either reselect or confirm the change. Upon confirmation, the system will automatically update and apply the new classification.
Yes. You can download results from SVOC through clicking the button 'download results to CSV' on the 'Classification Summary' page.
No. Users' personalized adjusted results are solely accessible to the account holder and not available to other users. This ensures both data security and preserves the integrity of each user's research.
No. All files uploaded by users along with any resultant output files are retained for a period of 24 hours. Upon expiration of this retention period, these files will be automatically purged from the server.
SVOC stands out due to its genome-wide coverage powered by expert-defined parameters, enabling systematic classification of both known and novel variants. It outperforms competitors and offers user-friendly features like high-throughput queries and interactive criteria adjustment, bridging the gap between theoretical frameworks and cancer research applications.
SVOC demonstrated optimal comprehensive performance in both accuracy and coverage on a benchmark set of 94 expert-curated variants. Its robust accuracy was further validated across multiple datasets, for both well-established variants and those lacking prior clinical interpretations. Specifically, it showed high concordance for oncogenic/likely oncogenic calls with OncoKB (96%) and ClinVar (89%), and strong agreement with functional categories from saturation genome editing of 6,959 BRCA2 mutations, achieving over 96% and 97% consistency for oncogenic and benign calls, respectively.
SVOC adheres to consensus parameters from prominent expert panels. These include ClinGen/CGC/VICC Guidelines Working Group, ClinGen’s Variant Curation Expert Panels (VCEP), the Somatic Variant Interpretation (SVI) Working Group, and Chinese expert panels.
Currently, users are not able to modify the parameters of SVOC online. Instead, since SVOC is open-source, advanced users with relevant technical capabilities can obtain the source code and deploy the SVOC tool on their own computing resources if they wish to explore custom configurations. Source code avaliable at: https://github.com/leqingsang/SVOC.
No. Currently, SVOC is specifically designed for and can only be used in the human genome. Our development efforts are focused on enhancing its performance and coverage for human somatic variants.
The thresholds, parameters, and functional data utilized in SVOC are tailored specifically for somatic variant classification. As such, SVOC is not designed for germline annotation, and we do not recommend its use for this purpose.
SVOC has not been validated in clinical scenarios. Therefore, we do not recommend using SVOC directly in clinical settings. If users intend to reference SVOC results for clinical decision-making, we highly recommend that they conduct a thorough manual review with the help of clinical and genomic experts.